算Fe的DOS
虚线:自旋向下;实线:自旋向上。
ToC
1. 参数测试
SYSTEM = Fe ISTART = 0 ICHARG = 2 ENCUT = 350 ISMEAR = 1; SIGMA = 0.2 ISPIN = 2; MAGMOM = 4 # save room LREAL=.FALSE. LWAVE=.FALSE. LCHARG=.FALSE.
Fe BCC 2.86 -0.5 0.5 0.5 0.5 -0.5 0.5 0.5 0.5 -0.5 1 C 0 0 0
Auto 0 G 9 9 9 0 0 0
PAW_PBE Fe 06Sep2000 …… …… …… ……
初始结构来自Material Project
ISMEAR = 1的设置是在官网看到的:
For relaxations in metals, use ISMEAR=1 or ISMEAR=2 and an appropriate SIGMA value (the entropy term should be less than 1 meV per atom). For metals a reasonable value is often SIGMA= 0.2 (which is the default). …… …… SIGMA should be as large as possible, while keeping the difference between the free energy and the total energy (i.e. the term entropy T*S) in the OUTCAR file negligible (1 meV/atom)
因此,还测试了一下SIGMA确保符合entropy T*S < 1 meV per atom的要求。
测试情况如下:
#!/bin/bash
n=1
for i in $(seq 275 25 600);
do
cp -r Fe/ Fe_encut_$n/
sed -i "/ENCUT/c\ENCUT = $i" Fe_encut_$n/INCAR
cd Fe_encut_$n/
qsub wz.pbs
cd ..
n=$((n+1))
done
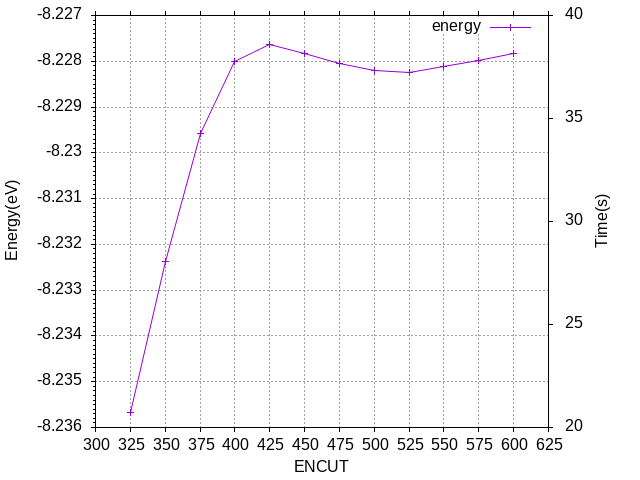
取ENCUT = 400即可。
#!/bin/bash
n=1
for i in 0.2 0.1 0.05 0.02 0.01;
do
cp -r Fe/ Fe_sigma_$n/
sed -i "/SIGMA/c\ISMEAR = 1; SIGMA = $i" Fe_sigma_$n/INCAR
cd Fe_sigma_$n/
qsub wz.pbs
cd ..
n=$((n+1))
done
| sigma | E(sigma->;0) /eV | EENTRO/eV | CPU time/s |
0.2 | -8.22801053 | -0.00276595 | 21.782 |
0.1 | -8.22766117 | -0.00054628 | 19.092 |
| 0.05 | -8.22761112 | -0.00023328 | 19.870 |
| 0.02 | -8.22756439 | 0.00002465 | 22.398 |
| 0.01 | -8.22758161 | 0.00002438 | 19.970 |
取SIGMA = 0.1即可。
#!/bin/bash
n=1
for i in $(seq 3 1 15);
do
cp -r Fe/ Fe_kp_$n/
cd Fe_kp_$n/
cat > KPOINTS <<HERE
Auto
0
G
$i $i $i
0 0 0
HERE
qsub wz.pbs
cd ..
n=$((n+1))
done
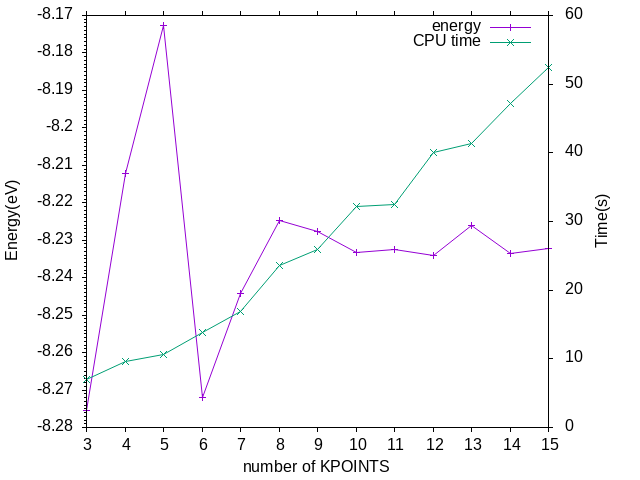
k点大小取11x11x11。
2. 结构优化
自动
设置了ISIF = 3,进行自动优化。
SYSTEM = Fe ISTART = 0 ICHARG = 2 ENCUT = 400 ISMEAR = 1; SIGMA = 0.1 ISPIN = 2; MAGMOM = 4 # move ISIF = 3 EDIFF = 1E-5 IBRION = 2 NSW = 50 POTIM = 0.1 EDIFFG = -0.01 # save room LREAL=.FALSE. LWAVE=.FALSE. LCHARG=.FALSE.
CONTCAR得到的立方晶胞大小为2.83258818459 A,Energy without entropy(sigma->0)= -8.23722355 eV
手动
#!/bin/bash
n=1
for i in $(seq 0.985 0.0005 1.005);
do
cp -r Fe/ Fe_$n/
cd Fe_$n/
cat > POSCAR <<HERE
Fe BCC
$i
-1.43 1.43 1.43
1.43 -1.43 1.43
1.43 1.43 -1.43
1
C
0 0 0
HERE
qsub wz.pbs
cd ..
n=$((n+1))
done
#!/bin/bash
n=1
for i in $(seq 2.826 0.001 2.836);
do
cp -r Fe/ Fe_$n/
cd Fe_$n/
cat > POSCAR <<HERE
Fe BCC
$i
-0.5 0.5 0.5
0.5 -0.5 0.5
0.5 0.5 -0.5
1
C
0 0 0
HERE
qsub wz.pbs
cd ..
n=$((n+1))
done
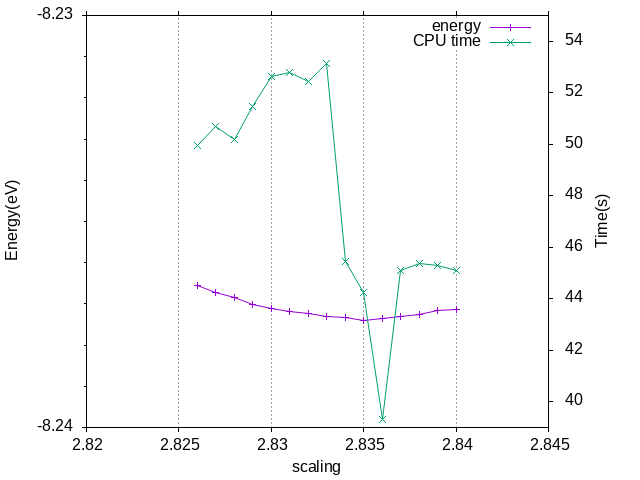
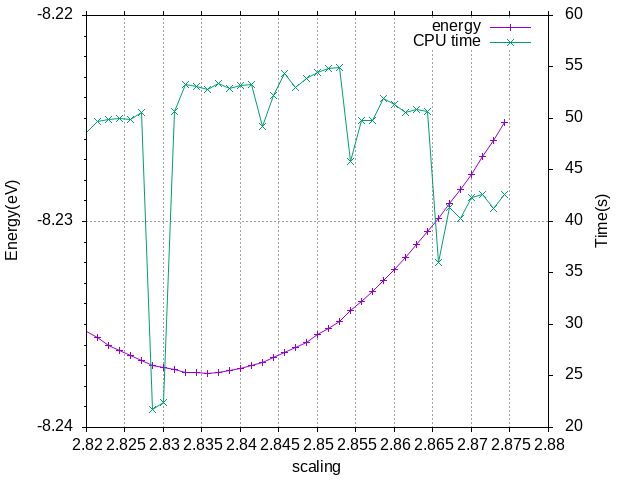
测试结果是取立方晶胞大小为2.835 A,Energy without entropy(sigma->0)= -8.23739980 eV,比自动的略小一点点
测磁性
用相同的晶胞(刚才手动测出来的那个)比较了三种磁性。
| INCAR | energy/eV | |
|---|---|---|
| PM | ISPIN = 1 |
-7.70467534 |
| FM | ISPIN = 2; MAGMOM = 4 4 |
-8.23575678 |
| AFM | ISPIN = 2; MAGMOM = 4 -4 |
-7.78285993 |
由上面的结果看出,体系是铁磁时能量最低,所以Fe的基态是铁磁性的。
3. 静态自洽
SYSTEM = Fe ISTART = 0 ICHARG = 2 ENCUT = 400 ISMEAR = 1; SIGMA = 0.1 ISPIN = 2; MAGMOM = 4 NCORE = 4
Fe BCC 2.835 -0.5 0.5 0.5 0.5 -0.5 0.5 0.5 0.5 -0.5 1 C 0 0 0
Auto 0 G 11 11 11 0 0 0
电荷密度文件CHG,CHGCAR拷贝到下一步非自洽计算。
4. 非自洽
DOS
SYSTEM = Fe ICHARG = 11 # DOS LORBIT = 11 NEDOS = 1409 ISMEAR = 1; SIGMA = 0.1 ENCUT = 400 ISPIN = 2; MAGMOM = 4 NCORE = 4
Auto 0 G 25 25 25 0 0 0
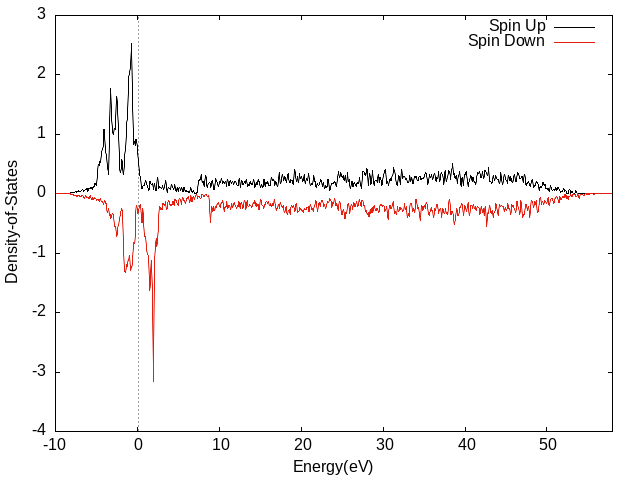
为了方便,先用vaspkit的111选项生成了TDOS.dat,再作图的。
1
2
3
4
5
6
7
8
9
10
set terminal png
set output 'DOS.png'
set xlabel "Energy(eV)"
set xrange [-10:58]
set xtics autofreq
set ylabel "Density-of-States"
set yrange [*:*]
set ytics autofreq
set arrow from 0, graph 0 to 0, graph 1 nohead lt 0
plot 'TDOS.dat' using 1:2 with lines title 'Spin Up' lt -1,'TDOS.dat' using 1:3 with lines title 'Spin Down' lt 7
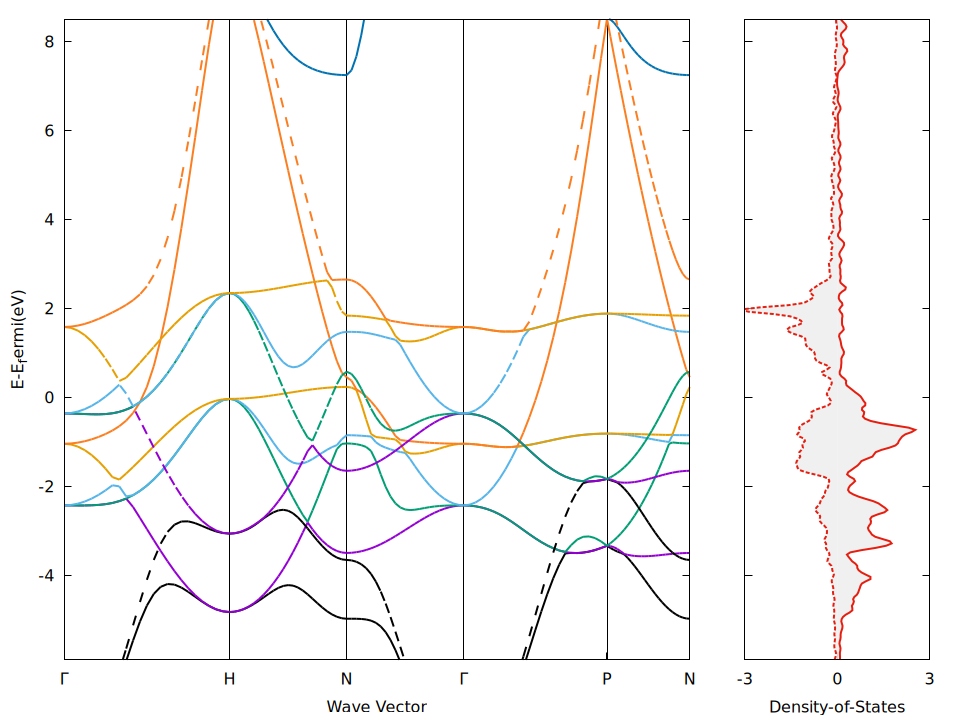
Band Structure
k points along high symmetry lines 25 line mode fractional 0 0 0 Γ 0.5 -0.5 0.5 H 0.5 -0.5 0.5 H 0 0 0.5 N 0 0 0.5 N 0 0 0 Γ 0 0 0 Γ 0.25 0.25 0.25 P 0.25 0.25 0.25 P 0 0 0.5 N
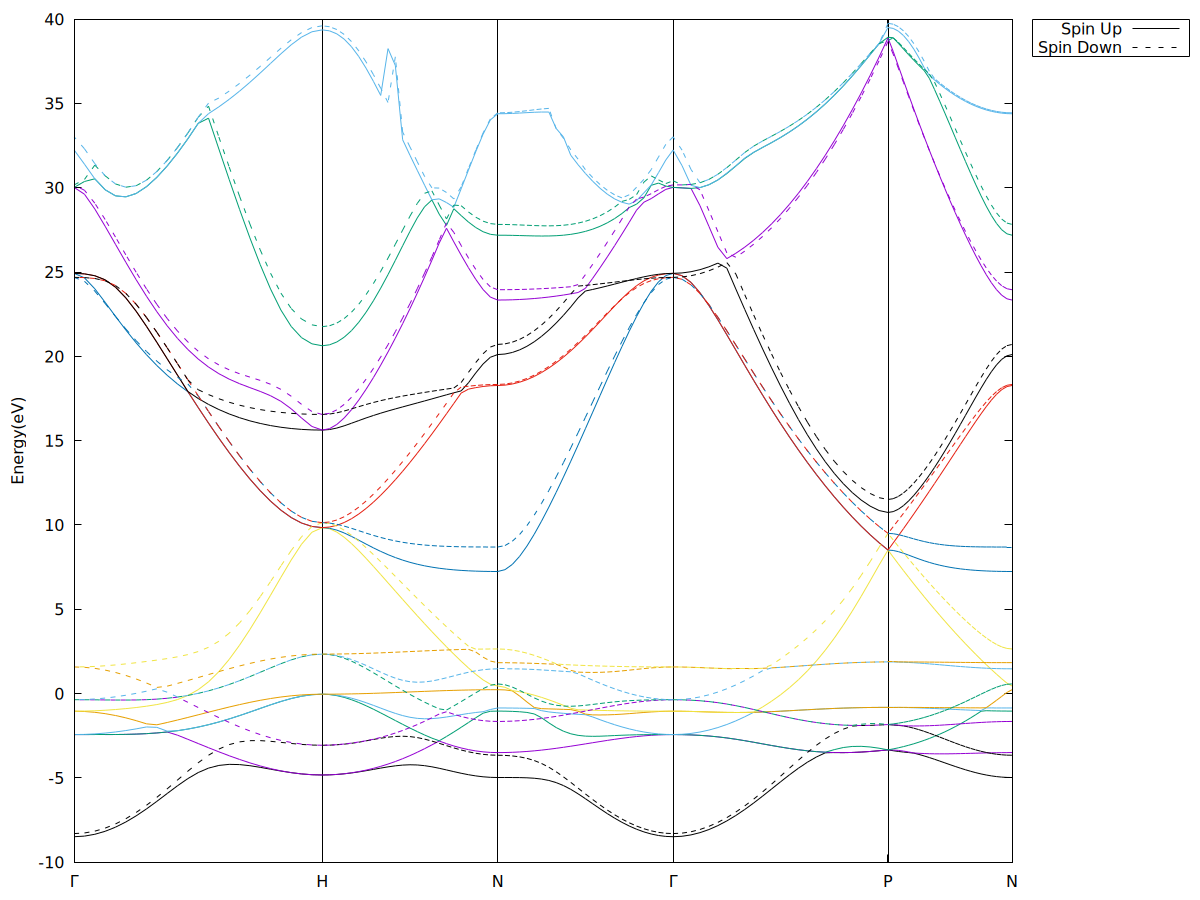
为了方便,先用vaspkit的211选项生成了BAND.dat,再作图的。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
set terminal pngcairo dashed size 1200,900
set output 'band-structure.png'
set xrange [*:*]
set xtics ("Γ" 0.000, "H" 2.216, "N" 3.783, "Γ" 5.351, "P" 7.270, "N" 8.378)
set ylabel "Energy(eV)"
set yrange [*:*]
set ytics autofreq
set arrow from 0.000, graph 0 to 0.000, graph 1 nohead lc rgb 'black'
set arrow from 2.216, graph 0 to 2.216, graph 1 nohead lc rgb 'black'
set arrow from 3.783, graph 0 to 3.783, graph 1 nohead lc rgb 'black'
set arrow from 5.351, graph 0 to 5.351, graph 1 nohead lc rgb 'black'
set arrow from 7.270, graph 0 to 7.270, graph 1 nohead lc rgb 'black'
set arrow from 8.378, graph 0 to 8.378, graph 1 nohead lc rgb 'black'
set key outside
set key box
plot 'BAND.dat' using 1:2:(column(-1)) with lines lc variable title "Spin Up", 'BAND.dat' using 1:3:(column(-1)) with lines dt 2 lc variable title "Spin Down"
5. 问题
- ENCUT和K点个数的测试叫做参数收敛性测试吗?目的是什么?计算精度到什么程度可以视为收敛?为什么先做这些测试再结构优化?结构优化之后还需要再测试一遍吗?
- 算DOS的时候官网这里推荐:“For the calculations of the DOS and very accurate total-energy calculations (no relaxation in metals), use the tetrahedron method (ISMEAR=-5)”于是跟着设置了, 但是后来把k点换成linemode算能带的时候,它显示不能用刚刚DOS的设置
ISMEAR = -5,所以为了保险我重新用原本的ISMEAR = 1算了DOS和能带,这样对吗?能带和DOS除了KPOINTS文件之外,其他参数需要保持一致吗? - PPT里在算非自洽的时候,设置了
ISTART = 1,这是为什么?我看到官网这里写“If the WAVECAR file is missing or if the WAVECAR file contains an inappropriate number of bands and/or k-points the flag ISTART will revert to ISTART=0.” 没有拷静态自洽的WAVECAR过去,这一项还需要设置吗? - PPT里算DOS的时候“设置能量间隔为0.05eV,NEDOS=1019”,NEDOS是指EMAX和EMIN能量间隔之间取的点的个数,可是这里不知道EMAX和EMIN,是怎么根据需求(能量间隔为0.05eV)得出NEDOS=1019的?我看了EMAX和EMIN的默认,比如EMIN是“default EMIN= lowest KS-eigenvalue - Δ”,这个从哪里能看到具体数值?EMAX和EMIN如果要手动设置的话,有什么基准吗?
- NEDOS选1019,1361,1501,2001的时候,DOS的峰不一样高,是因为调整间隔之后算的点不一样?
6. 反馈
- 测磁部分,INCAR中MAGMOM后面的atomic spin moment与POSCAR中原子数对应。换言之,测AFM时至少POSCAR里要含2个Fe
- BAND structure中如果不需要轨道投影的话,只需要spin up用红色,spin dwn用黑色就行了,如果需要看分轨道的能带结构,则记得标注 s– p– d–
- 根据你测试的能量收敛情况可以看出,不同ENCUT和K对最终体系优化收敛速度和能量精度都有较明显的影响。这个有点一体两面的意思,ENCUT和K越大,计算越消耗资源,收敛结果相对越好(优化结构越理想,体系能量越低),测试主要目的是找到一个折中点。 测试一般是在大量系统性计算开始前,开始结构优化后就不用再测试了。
- 非自洽计算能带结构时,确实不能用tetrahedron method,我一般用ISMEAR=0,>0也没问题
- 没有拷贝WAVECAR到静态非自洽计算目录的话,INCAR里设置ISTART=任何值,效果都是一样的。
- NEDOS一般是在静态计算时设置的,在此之前的优化计算过程中会产生DOSCAR文件,在设置NEDOS之前,可以先查看一下之前优化计算时DOSCAR的头部行,那里有EMAX和EMIN的范围;DOSCAR中第6行分别是:EMAX, EMIN, NEDOS(默认301), Fermi Energy;你在静态计算时,可以在INCAR里自己定义EMIN和EMAX,然后两者之间能量范围/能量间隔(如0.05eV)=你要设置的NEDOS值,不手动设置EMIN和EMAX,则非自洽计算取的能量范围就是之前计算的EMIN,EMAX;±3eV差不多吧,不过这个还以实际情况为准,而且能量范围未必取费米面为中点,费米面以上太高能量区域意义也不大。
- NEDOS可以视作撒取样点,能量间隔不同,点的疏密度不同,dos峰绝对高度会有所不同的。DOS的绝对值意义不大,主要看分布。当然DOS的积分是有实际意义的