CrCuSe2块材和不同单层结构
ToC
Bulk
1.1 参数设置
以下是一些共通参数:
ENCUT = 520
EDIFF = 1E-6
ISMEAR = 0; SIGMA = 0.05
ISIF = 3
IBRION = 2
POTIM = 0.1
EDIFFG = -0.01
NELM = 100
NSW = 99
LREAL =Auto
LASPH =.TRUE.
打开LASPH是因为看到wiki写了”This is essential for accurate total energies and band structure calculations for f-elements (e.g. ceria), all 3d-elements (transition metal oxides), and magnetic atoms in the 2nd row (B-F atom)”
##kpoints
Auto
0
G
7 7 7
0 0 0
##potcar
.54 PAW: Cr_pv Cu_pv Se
原胞是三方的不方便, 所以采用了包含12个原子的六方晶胞作为unit cell
Cr3 Cu3 Se6 1.0 1.8742218411589515 -3.2462474535425900 0.0000000000000000 1.8742218411589515 3.2462474535425900 0.0000000000000000 0.0000000000000000 0.0000000000000000 19.1064188631023697 Cr Cu Se 3 3 6 direct 0.0000000000000000 0.0000000000000000 0.0006430000000000 Cr3+ 0.6666666666666666 0.3333333333333333 0.3339763333333333 Cr3+ 0.3333333333333333 0.6666666666666666 0.6673096666666667 Cr3+ 0.6666666666666666 0.3333333333333333 0.1905393333333333 Cu+ 0.3333333333333333 0.6666666666666666 0.5238726666666667 Cu+ 0.0000000000000000 0.0000000000000000 0.8572059999999999 Cu+ 0.6666666666666666 0.3333333333333333 0.0680023333333334 Se2- 0.0000000000000000 0.0000000000000000 0.2554820000000000 Se2- 0.3333333333333333 0.6666666666666666 0.4013356666666667 Se2- 0.6666666666666666 0.3333333333333333 0.5888153333333334 Se2- 0.0000000000000000 0.0000000000000000 0.7346690000000000 Se2- 0.3333333333333333 0.6666666666666666 0.9221486666666667 Se2-
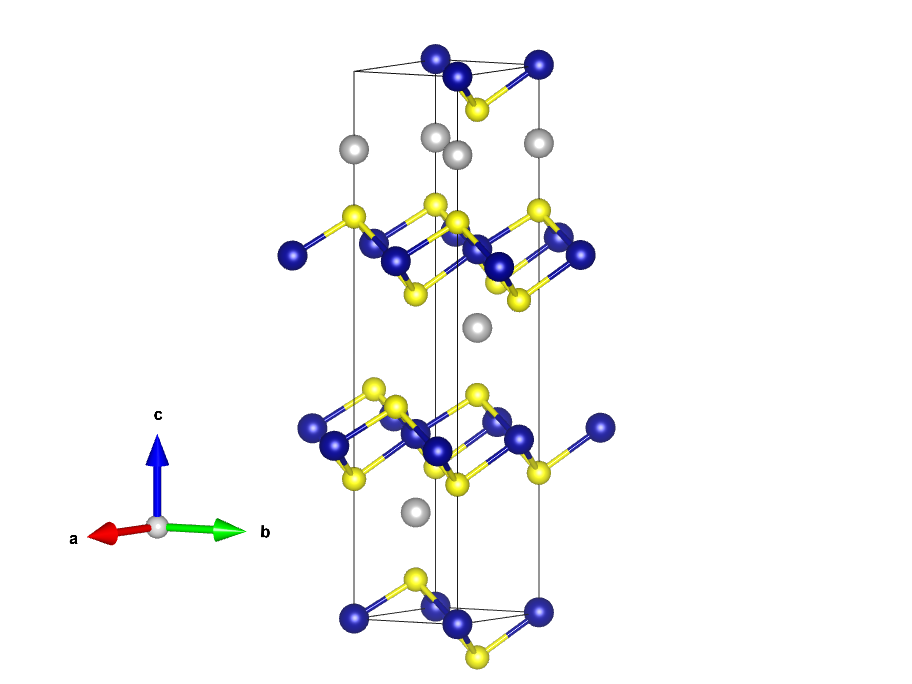
FM的POSCAR直接用该unit cell, 对应MAGMOM的Cr部分设置了3*3.0,Cu和Se部分设置了3*0.1 6*-0.1
层间AFM的POSCAR用了1x1x2的supercell,对应MAGMOM的Cr部分设置了3.0 -3.0 3.0 -3.0 3.0 -3.0,Cu和Se部分6*0.1 12*-0.1
层内AFM的POSCAR用了2x2x1的supercell,对应MAGMOM的Cr部分设置了3.0 -3.0 3.0 -3.0 3.0 -3.0 3.0 -3.0 3.0 -3.0 3.0 -3.0,Cu和Se部分12*0.1 24*-0.1
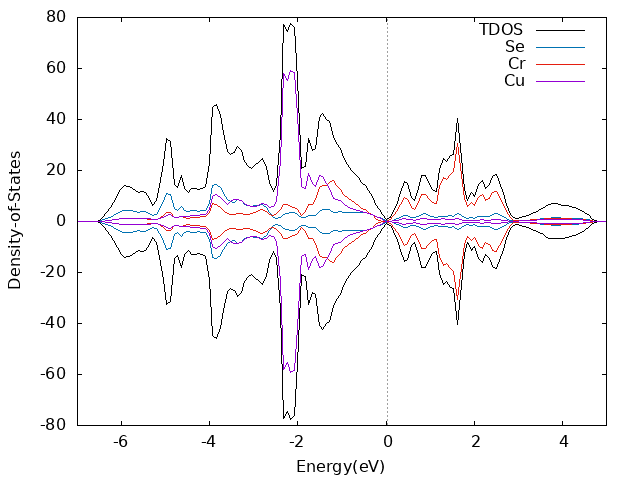
算dos的时候kpoint设置的13x13x13,NEDOS设置的1019
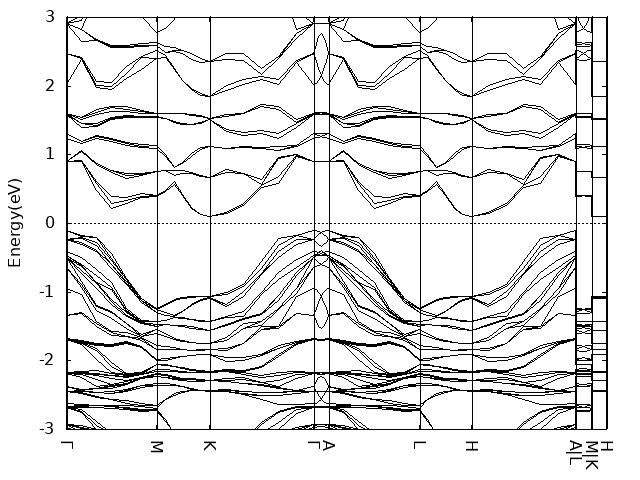
算band的时候kpoints along high symmetry lines设置的7(额,好像有点小),路径用的是这篇的图十三标注的Γ–M–K–Γ–A–L–H–A|L–M|K–H。
- 其中,对于supercell的高对称点,分数坐标乘上相应倍数。例如,1x1x2的supercell,其x和y方向的逆格矢不变,z方向逆格矢是原来的二分之一,用原本逆格矢写出的z方向坐标如果是0.5,那么用新的逆格矢写出的z方向坐标应该是1
1.2 结果
| Mag. | ∆E(meV/unit cell) |
|---|---|
| FM | 0 |
| Inter-AFM | -4.9 |
| Intra-AFM | 110.2 |
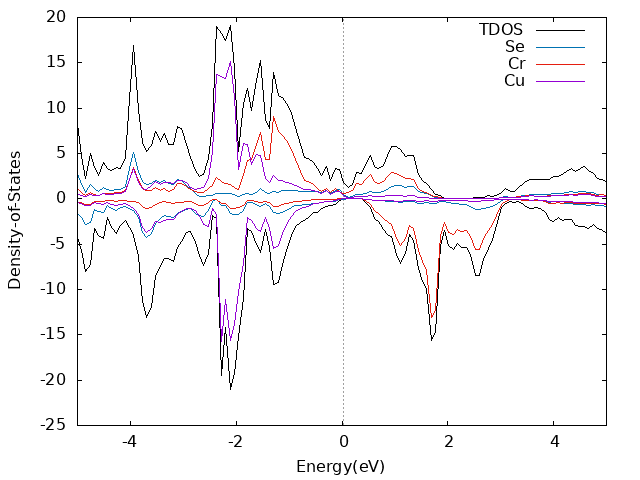
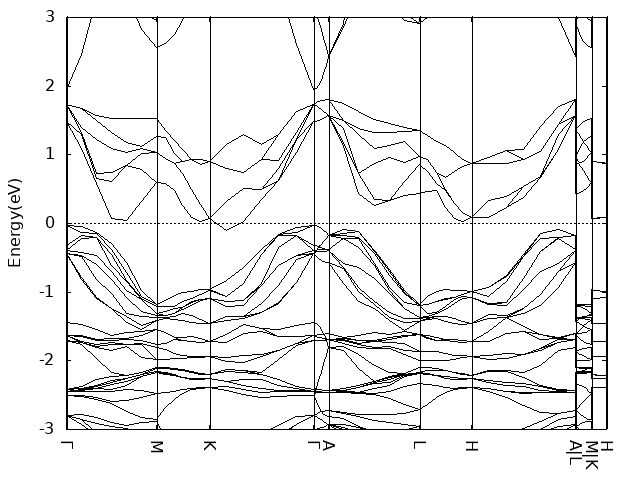
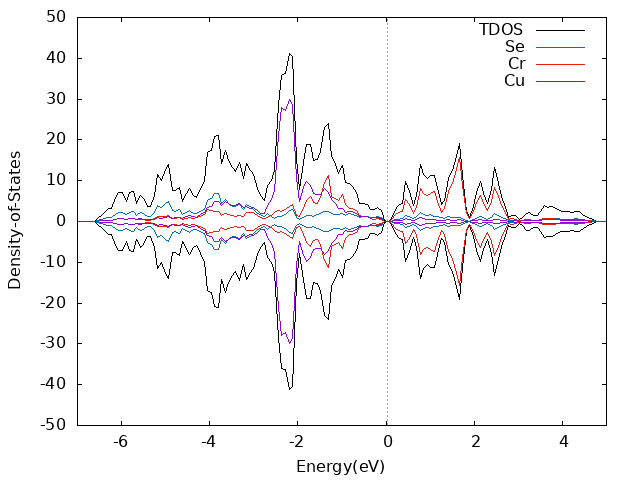
Monolayer
2.1 参数设置:
共通参数:
ENCUT = 520
EDIFF = 1E-6
ISMEAR = 0; SIGMA = 0.05
ISIF = 4
IBRION = 2
POTIM = 0.1
EDIFFG = -0.01
NELM = 100
NSW = 99
LASPH =.TRUE.
##kpoints
Auto
0
G
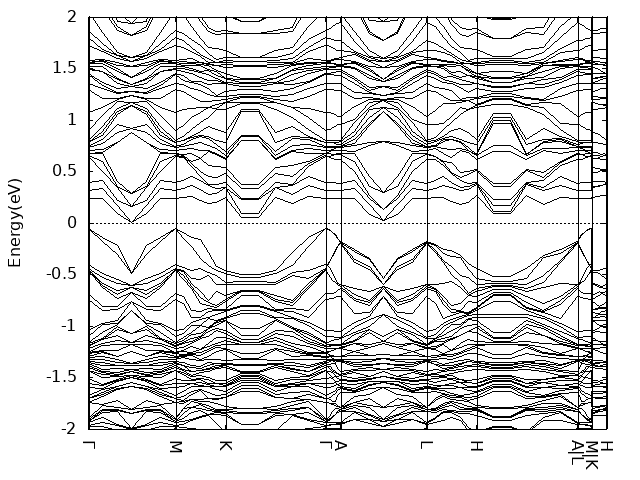
13 13 1
0 0 0
对于POSCAR, 先从块材unit cell中选取单层,再调整原子的位置如下图:
Cr3 Cu3 Se6 1.0 1.8742223557 -3.2462476161 0.0000000000 1.8742217247 3.2462479804 0.0000000000 0.0000000000 0.0000000000 40.0 Cr Cu Se 2 1 4 Cartesian 1.874222145 -1.082084581 6.381085262 1.874221935 1.082084946 12.749904202 1.874221935 1.082084946 10.009336836 0.000000000 0.000000000 4.881346039 1.874221935 1.082084946 7.668093619 1.874222145 -1.082084581 11.250145874 0.000000000 0.000000000 14.036893454
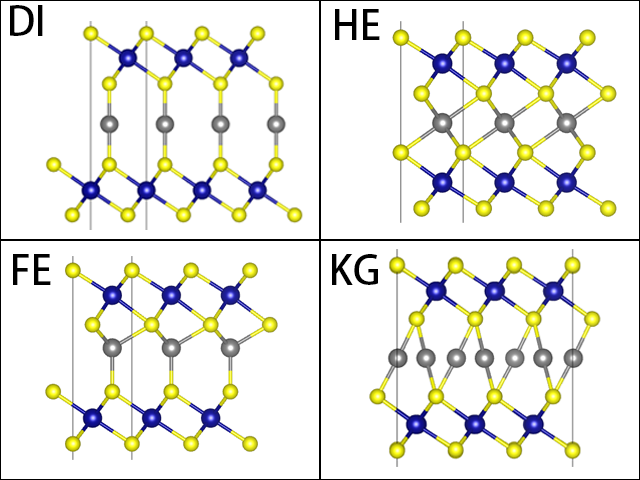
- DI 将两个Cr-Se层的Se对齐,在对齐的Se的中央放Cu
- FE 和块材相同,不调整位置
- HE 将两个Cr-Se层的Cr对齐,在对齐的Cr的中央放Cu
- KG 将单层扩大到3x3x1,隐藏Cr、最上层和最下层的Se,从C轴看可见Se形成了很多六边形,在六边形的间隔开的三边的中点放Cu,如图
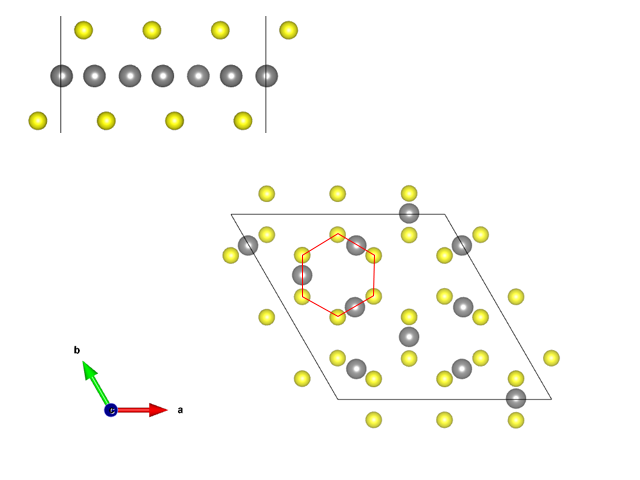
调整后再统一将晶胞的z方向长度改为40
MAGMOM统一给Cr设置的3,Cu和Se设置了-0.1,对KG额外设置了LREAL=Auto和SYMPREC=1E-4
2.2 结果
| FE | HE | DI | KG | |
|---|---|---|---|---|
| d(Å) | 3.57 | 3.00 | 4.55 | 4.12 |
| |z|(Å) | 42.33 | 42.02 | 43.58 | 43.71 |
| ∆E(meV) | 0 | 185.4 | 275.8 | 266.6 |
d是夹住Cu的两层Se之间的距离,|z|是晶胞z方向垂直距离
问题反馈
- bulk验算中,INCAR里的LASPH一般在DFT+U、HSE等情况下需要设置,普通DFT加了也不会有问题;
- bulk磁基态验算中,Inter-AFM(i.e.,A-AFM)结构用了1×1×2的超胞,但是我用你六方unitcel的POSCAR扩胞112后,如果设置Cr的磁性为 3 -3 3 -3 3 -3,磁序成了近邻六方unitcell之间AFM,unitcell内部FM了。不知道是不是你扩胞后的POSCAR和我的不同导致的。另外,Cu和Se上的局域磁矩很小,你在INCAR初始设置时直接设置为0就行
- band计算时注意设置LMAXMIX参数,否则很容易出现dos和band不能对应的问题,例如你计算的bulk磁基态inter-AFM,dos上看0~2eV范围内没有gap,但是band显示在靠近2eV处存在小带隙。类似CuCrSe2之类层状六方结构,后续band可以只设置Γ-M-K-Γ就可以了。尤其是后续单层结构计算时。
- 关于bulk CuCrSe2 可以边算边调研相关文献,看看实验报道或者其他理论研究有没有与你目前验算结果相印证或者不一致的。
- 加了真空层的单层结构,在全局优化的时候需要采用 vasp_std_adir配合OPTCELL使用,限制晶胞在真空层方向的参数优化,不然全局优化后z方向晶格参数就不是一开始统一的40A了,这里你优化后晶胞畸变不厉害,有的会膨胀或收缩的特别夸张,导致后续没法算了。关于OPTCELL配合vasp_std_adir的使用,你试试看,不难,服务器上基本都有编译好的可执行程序。
- 不用重新优化了,就拿你目前isif=4优化收敛的单层结构,重新修改统一c=40A,然后放开ab,固定c,进行全局优化再看看。你有没有发现,从bulk剥离成单层后,每个单层内有两个Cr离子层,中间夹着一个Cu离子层,你再看一下剥离后单层的化学式,这里Cr上应该有变价。不含磁优化找到能量最低的单层结构后,再试试FM,inter-AFM,还有intra-FM,前两者应该能量近似相同,主要是判断一下单层,到底是面内FM还是面内AFM能量低,估计CrSe2面内FM可能性比较大